Choosing the right safety reporting workflow for your study

Safety comes first. Always.
Especially in the clinical setting. Naturally and importantly, safety event reporting and pharmacovigilance are subject to severe regulatory oversight.
Consequently – and here, I’m confident you will agree with me based on your own experience: When planning a clinical project, one of the most sensitive, and coincidentally most important issues, is the establishment of a secure, feasible, functional and effective safety reporting workflow.
But how can you make sure you’re choosing the right safety reporting workflow for your study?
We will discuss the characteristics of 3 common safety reporting strategies in this article. By the end, you will know their SWOTs (strengths, weaknesses, opportunities, threats). This gives you a good starting point or perhaps some new ideas for choosing your optimal approach.
But let me start by explaining why the decision for a notification process in your study should be considered carefully.
4 demands upon safety reporting today:
No matter if your project is of interventional or observational kind – meeting these demands with the right safety reporting workflow is a challenge.
A challenge we will take up by the example of a generic multi-national study with the reporting language English and with involvement of a CRO, a local pharmacovigilance department and a global pharmacovigilance database.
With the help of this example, we will work ourselves through 3 common notification processes: “paper2paper”, “hybrid” and “the future is now”.
Workflow 1: “paper2paper” – the traditional workflow
This is the classical workflow that has been well established in clinical research before the whole digitalization process began. “Paper2paper” describes that both, clinical data and AEs, serious and non-serious, make use of a paper-based report form.
Old-fashioned you say? Maybe. But the workflow also holds some strong advantages: its’ simplicity and the physician’s familiarity with the process being only two of them.
— What exactly is the process behind?
As the name says – paper is added to paper: Clinical data are recorded on paper case report forms (CRF). And SAEs are reported by the physician by means of a paper SAE report form that is usually faxed to the CRO.
“Paper CRF + paper SAE report form”
The CRO processes this SAE information (e.g. check for completeness, plausibility, duplicates, events of special interest etc.) and, if necessary, initiates a follow-up and forwards the SAE to the local affiliate. Here, the information is further processed and finally entered into to the global pharmacovigilance database.
Of course, the CRO may also be tasked with further services (like data entry into the pharmacovigilance data base). However, for the here described workflow of obtaining information from the sites, this doesn’t play a major role.
Within the CRF, the investigator documents all AEs (non-serious AEs and SAEs) on the provided AE pages which, later on, are entered into the clinical database.
— Where are the challenges?
By legal requirements, it may no longer be sufficient to only summarize the non-serious AEs in the study report. For clinical trials, the novel European clinical trial regulation rather stipulates that mechanisms should be in place allowing for timely reporting of all events that are relevant for benefit-risk balance evaluation (REGULATION (EU) No 536/2014).
“Reporting of ALL events relevant for benefit-risk balance evaluation”
The sponsor of a clinical trial, thus, should be able to check all safety-relevant events in a timely manner.
In practice this means that, for example, all documented non-serious AEs should regularly be screened for accumulation of certain events.
For individual case safety reports and solicited reports from post-marketing studies (e.g. non/interventional studies) on the other hand, all non-serious adverse reactions shall be reported to the regulatory bodies of the respective EU member states within 90 days of their occurrence (Directive 2010/84/EU).
Therefore, in many cases the safety reporting workflow will also need to incorporate a step for reporting all non-serious AEs to the CRO. The sites’ readiness for this might be the limiting factor.
— Solutions and Considerations:
In my experience, the most straight forward approach to face this regulatory challenge is to prompt that all AEs, regardless of seriousness, must be reported to the CRO within 24 hours.
At the CRO, non-serious AEs are being checked for completeness as well as general and sponsor-specific seriousness criteria and summarized in aggregate listings afterwards.
The resulting aggregate reports are then forwarded as line listings to the local pharmacovigilance department for further processing and referring to the global pharmacovigilance department for regulatory reporting.
“For this scenario, it is crucial to provide the sites with support wherever possible.”
After all, the success of the “paper2paper” workflow (and of your study altogether) highly depends on the sites’ cooperation. Therefore, user-friendliness of study materials and support wherever possible are crucial.
For example, AE pages of the CRF should be easily removable and re-importable into the site file.
Also, provide sufficient guidance for the sites on ”what to do when”: In case of one or several non-serious AEs, only the AE page must be faxed to the CRO; in case of an SAE, the AE page as well as the SAE form need to be transferred.
Keep the required information on the AE page as minimal as possible to avoid excessive documentation efforts for the sites: In most cases, the reported term, a start and a stop date, and the seriousness and causality assessment should suffice.
“The CRO serves as a filter and control body.”
On the other side of the chain in the “paper2paper” scenario you have the CRO which serves as a filter and control body. The process provides that all AEs reach the CRO within 24 hours.
Your CRO must then ensure that
You see that it is absolutely crucial to identify upfront which kind of information is essential for the non-serious AE processing.
Therefore, I strongly recommend consulting the local and/or global pharmacovigilance department already during the development of your CRF.
“The reporting modalities are dependent on the time point zero of awareness.”
During the development phase of your CRF also always keep in mind: The more information required by the sites regarding non-serious AEs, the lower the sites’ motivation to report the information on time and the greater the amount of time and effort required for the recipient (as the CRO will need to check – and perhaps query – more items).
The reporting modalities for your workflow are strongly dependent on your definition of the time point zero of awareness. Most sponsors will choose the conservative approach described above – is this true for your company as well?
— One last remark:
In some cases, e.g. for studies with short duration per patient, or studies with regular monitoring visits, it might be sufficient to only subject SAEs to the expedited reporting.
For this approach, the site is required to send the CRF/CRF parts containing the AE information to the CRO immediately after the patient completes the study or after the respective monitoring visit. Immediately upon receiving the documents from the sites, the CRO then lists the non-serious AEs.
This approach can be more time saving and more user friendly, however, might lead to bottlenecks in data entry and processing at the CRO and the local and/or global pharmacovigilance database. The estimated AE numbers and study design modalities will need to be considered carefully before choosing this option.
Now let’s embrace the remarkable opportunities that digitalization has given us and move to our second possibility for a notification process: the “hybrid scenario”.
Workflow 2: The hybrid scenario – eCRF with paper-SAE reporting
This is the classical workflow when using an electronic CRF (eCRF) instead of a paper one. The “hybrid” consists of an online recording of clinical data combined with a paper-based SAE reporting.
— What exactly is the process behind?
All clinical data, including AE/SAE information, are collected online in the eCRF. However, SAEs must additionally be reported via paper forms (e.g. fax) to the CRO, promptly after the site’s awareness.
“eCRF + paper SAE report form”
Like in the “paper2paper” scenario, the CRO processes the SAE information and forwards it to the local pharmacovigilance department for further processing and referral to the global pharmacovigilance department.
And also in the same way as in workflow 1, queries regarding SAEs are sent by/via the CRO to the sites offline (i.e. via fax).
You can see, we are talking about an online-offline combinational approach here. The strengths of an online eCRF with its possibilities for automation are supplemented by the familiar offline paper-based reporting.
One major benefit of this online-offline combination is that all AE data, i.e. non serious AEs and SAEs, are entered into the eCRF and are available for generating aggregate reports (e.g. non serious AE line listings) at any time as soon as the site enters that data.
Of course, edit checks and constrains that guide the investigator during the data entry process are helpful tools, not only for clinical data recording but also for safety reporting – one big pro for an eCRF-based solution!
— Where are the challenges?
We all know that the reality of clinical routine does not always meet our hypothetical expectations of a process. In many cases, data is entered into the eCRF with a great latency after the patient has been seen.
“Discrepancies between pharmacovigilance and clinical database”
What happens is that the information about an SAE can differ substantially between the pharmacovigilance database and the clinical database at a given time – not only by its entry date but also by its informational content.
However, most sponsors define their time point zero of awareness as “the moment when potential (S)AE data are entered into the eCRF”.
If you have received the corresponding fax already, you are safe. However if the site forgets to send the fax, you have to make sure that as soon as you reach that moment of data entry into the eCRF, you also get informed about it.
Of course, a good eCRF can do this job for you. It is however crucial to define competent control and alert mechanisms and workflows (see also figure 1: SAE reporting scenarios).
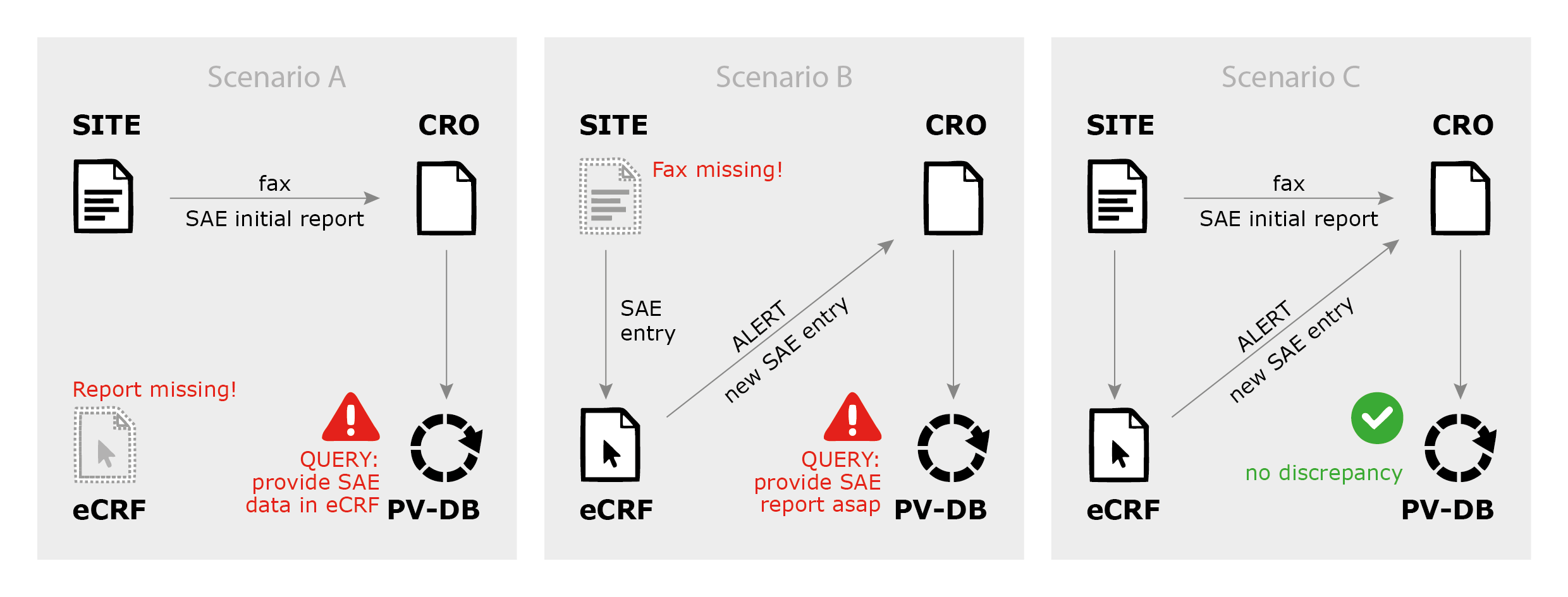
— Solutions and Considerations:
In order to ensure a consistent safety reporting, the CRO as your filter and control body has to ensure that all eCRF entries are checked for hidden/unreported AEs/SAEs or, if applicable, for events of special interest according to criteria defined in the protocol.
The easiest way to do so is to trigger an alert whenever relevant AE data are entered into the eCRF and to perform regular checks of all free-text entries, where alerts are not possible or useful.
“Keep line listings concise and their volumes manageable.”
Utilizing customized eCRF reports can allow you for on-demand aggregate reporting of AEs (e.g. line listings). In that way you can have a clear view of all AEs occurring in your study at any time.
The structure and content of these on-demand reports need to be clearly pre-defined, preferably before the eCRF has gone live.
They can, for example, also contain information derived from any of the forms of the eCRF (e.g. medical history, concomitant medication etc.).
However, consider carefully which structure of the listings you want to use in order to keep your line listings concise and their volumes manageable. Otherwise, you might miss the wood for the trees (see figure 2: AE line listings on patient vs. event level).
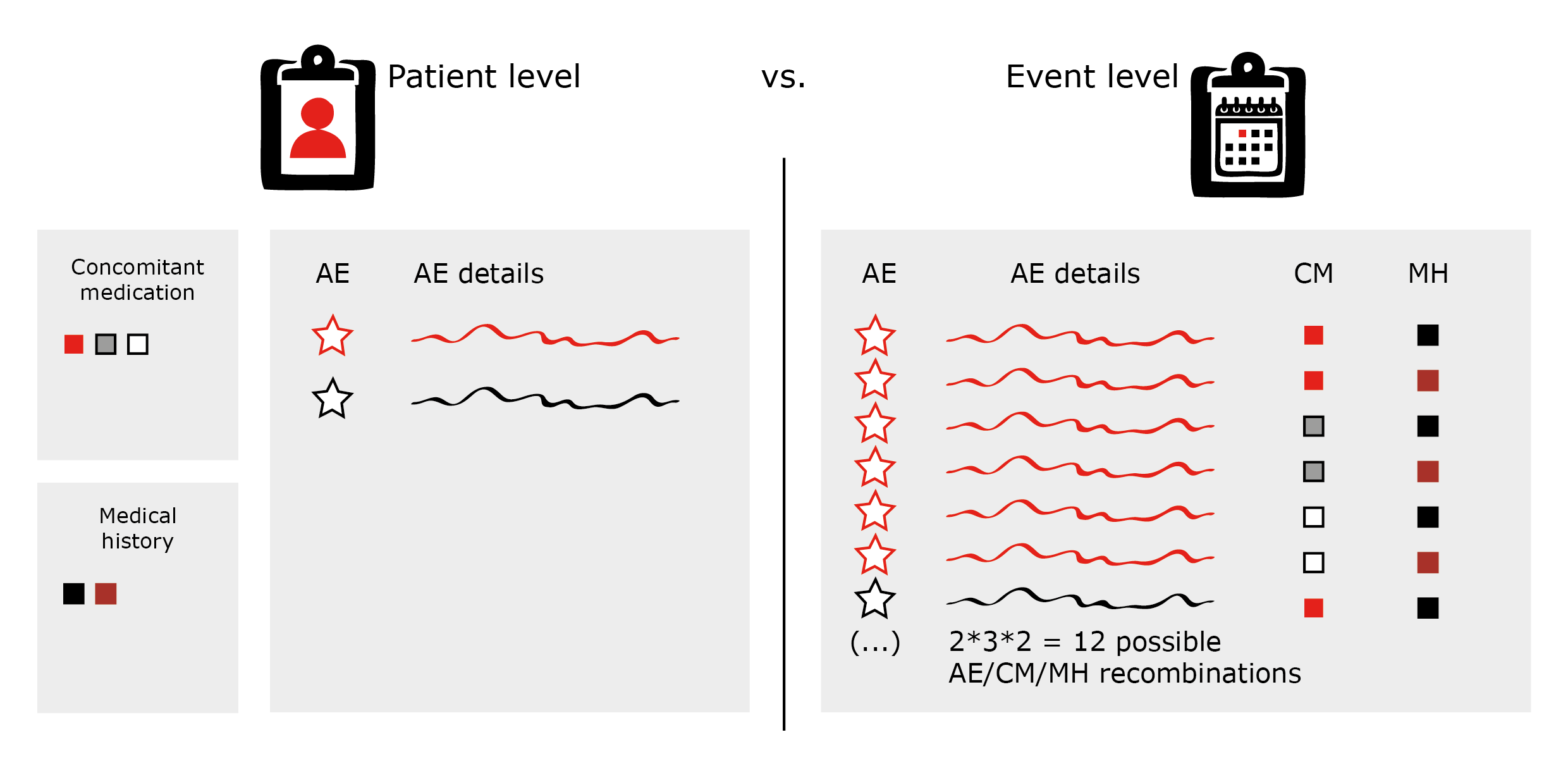
As mentioned before, the latency between the SAE report being sent by fax and the information being entered into the clinical database (eCRF) can lead to some confusion; especially if the AE term is not equal or similar enough.
In most cases, I recommend to trigger email alerts whenever an SAE is entered into the eCRF; but the CRO must ensure that this report has already been reported by fax, and if not, initiate the query process (see above, figure 1, scenario B: fax is missing).
“Regular reconciliation is almost a must.”
A field in the eCRF, where the date when the paper report was faxed to the CRO is documented, can be a helpful tool here. However, this does not allow for omitting to track and to compare the SAE information received via fax and via eCRF alert. The engagement of a trained CRO staff member is advisable for this important task.
Make sure that it is clearly defined how to proceed in the case follow up information is provided (e.g. shall the initial report date be updated to the date when the follow-up was sent?).
Particularly because the “hybrid” safety reporting workflow is a mixture of online and offline tools that can lead to complexity, regular reconciliation is almost a must – at least to ensure that all data in the eCRF have been processed and sent to the safety database. (see above figure 2: SAE data reconciliation).
— One last remark:
To conclude, a general remark affecting both above paper-based reporting workflows:
At least in Germany, but likely in all European countries, the telephone network providers are in the process of updating their fax protocols.
Even today, this causes problems in the transmission of faxes from older models (as often used in smaller sites and hospitals) to the CROs and/or local pharmacovigilance departments (where usually the most recent technologies are implemented).
Therefore, ensure to provide alternative reporting possibilities (e.g. provide an email address of the CRO/local pharmacovigilance department) or consider allowing for email reporting rather than fax altogether.
But bear in mind your company’s SOPs and current local laws (not only yours but also the sites’) regarding data protection and ensure time for establishing secure email transmission between all parties involved.
In addition, make sure to implement a regulation for receipt confirmation when using email for SAE reporting.
Finally, I recommend providing your CRO with the full contact details for all sites a priori so that in case of technical difficulties the CRO can contact the site in a timely manner.
After hearing about the above two safety reporting workflow possibilities, your logical conclusive question might be, why involve paper at all? Why not switch to fully digital SAE reporting altogether? This brings us to workflow 3.
Workflow 3: The future is now – electronic SAE reporting via the eCRF
And indeed – safety reporting in clinical and non-interventional studies is currently at a starting point of implementing eSAE solutions into the eCRF.
The idea behind this is fairly simple: why ask the physician to document one and the same event on different places when all information is available in the eCRF anyhow?
— What exactly is the process behind?
The only contrast to the “hybrid” scenario above is that safety data are only entered into the eCRF.
When a SAE is documented, the SAE data, along with relevant data on demography, medical history, etc. are automatically provided on an electronic report (usually PDF format) that is then sent to the CRO by email.
“How cool is that?” you might say. And I agree, there are many benefits to this approach: automation, standardization and accessibility being named first.
However, careful considerations during the set-up phase of this notification process should address internal challenges (applying to the sponsor’s own SOPs) as well as external challenges (demanding for a precise workflow between the EDC vendor, sites, monitoring staff, the CRO and the local pharmacovigilance department).
Let’s have a look at what these challenges are.
— Challenges and Solutions:
Only relevant information please!
This challenge addresses the problem of too much information at the wrong place. Let me explain by an example of a regularly used SAE form section.
The physician is asked to provide the additional information that he/she renders important for the case (i.e. “please provide relevant medical history”). Your experience might let you agree that in the vast majority of cases this field is left untouched.
However, if all medical history is automatically “thrown” from the eCRF into the SAE report, without the physician’s assessment of its relevance for the case, the sponsor’s assessment of the case might be wrongly impacted.
For example: A site reports an SAE “gamma GT increased” within a clinical study. The patient’s medical history lists “diabetes, ongoing since 1999”, “hyperlipidemia, ongoing since 1999” and “alcohol abuse between 1980 and 1985”.
“Offer the physician a choice”
If the workflow does not explicitly ask the physician to provide the relevant medical history and, instead, all of the entries are placed in the eSAE report’s medical history field, this will make the sponsor’s causality and expectedness assessment rather difficult.
Perhaps, on a paper form the physician might have deliberately not mentioned the patient’s alcohol abuse from more than 20 years ago as it is might be completely irrelevant for the case.
Thus, I recommend technical options, where the physician is offered a choice (e.g. a drag and drop menu) if he/she considers a given entry relevant for the case or not.
Additionally, it should be clearly visible on the final output, if the information is all medical history there is, or if it is medical history selectively marked as relevant by the physician.
Who is pulling the trigger?
A precise action needs to be defined that shall trigger the sending of the eSAE report.
For some systems, the report is sent when the reporter clicks on the “save” button to save his/her entries; but careful – some people save after every entry they do, leading to massive email traffic.
“Avoid massive email traffick and incomplete reports”
Some systems trigger the report without the reporter knowing (i.e. when the entry is interrupted for longer than a pre-defined time limit), which might also cause a flood of incomplete reports.
And yet again some systems allow for triggering the sending when a digital signature is placed. This might however be problematic as often only investigators/physicians have the right to sign off eCRF pages. In turn, a study nurse who does not have the right could not report an SAE.
Keep at least some paper!
Be aware – you are working on the basis of an online tool with all its technical issues there are.
It is crucially important to consider ways to ensure on-time reporting of SAEs in cases where the eCRF is not functional, or the investigator has lost/forgotten his/her log in information.
“Offer alternative reporting possibilities”
Thus, the implementation of an eSAE workflow will always need to include the opportunity for a paper SAE report to be faxed or emailed, should it be necessary.
eSAE is tasty, but too many cooks spoil the broth!
On the CRO and sponsor side make sure to carefully and precisely define who shall receive which information.
Just because the system can distribute eSAE reports to any multitude of recipients, it does not mean that this is practical or meaningful.
“Define a hierarchical approach”
It is therefore important to define a hierarchical approach.
For instance, the CRO can serve as the first point of contact in order to filter the reports (i.e. for errors or incompleteness). This ensures that only consolidated, complete, or at least valid reports are forwarded to the local pharmacovigilance department within reasonable time frames.
Keep in mind – it is still a CRF!
And as such, it has effects on the results of your study.
Therefore, be always clear to what degree the pharmacovigilance process can have an impact on your study endpoints.
“Be aware of effects on your study”
For instance, I advise you to carefully examine if pharmacovigilance related queries (or data entries/corrections) can affect your endpoints. Just keep the data management team and even biostatisticians involved in the set-up process.
For international studies I additionally recommend to provide that the safety query process is the same for all countries involved, as otherwise a country-specific bias might be introduced to relevant study endpoints.
— Considerations:
Although eCRFs have been around now for quite a while, eSAE reporting is yet to become the reporting standard.
The technical possibilities are growing continuously. Today, we are in the position to generate high-quality SAE reports where, based on audit trail information, relevant new information can automatically be highlighted and reported – all without printing one page of paper.
The greater the technical possibilities, the more important it is, from my experience, to define a workflow where all involved parties know their roles and responsibilities.
And to do this a priori and with enough time for all concerned parties to think profoundly through the process.
As usual with the eCRF: almost anything is possible in the beginning, but once established, changes are not advisable.
Picture: @PureSolution /Fotolia.com



