FAQ zur Klinischen Prüfung mit Medizinprodukten

Viele Hersteller werden vor eine große Herausforderung gestellt, wenn sich im Rahmen der klinischen Bewertung über den Literaturweg herausstellt, dass nicht ausreichend klinische Daten zur Verfügung stehen, um die Konformität mit den Grundlegenden Anforderungen (EU RL 93/42EWG, Anhang I) zu zeigen und somit eine klinische Prüfung erforderlich wird.
Häufig besteht eine große Unsicherheit bezüglich der Regularien für klinische Prüfungen mit Medizinprodukten sowie Unklarheit darüber, für welche Art von Medizinproduktestudien welche Art von Genehmigungen und Voten vorliegen müssen.
Um Ihnen den Einstieg in die klinische Prüfung mit Medizinprodukten zu erleichtern, haben wir für Sie im Folgenden einige wichtige Fragen und Antworten zu diesem Thema zusammengestellt.
Klinische Prüfungen von Medizinprodukten im Sinne des Medizinproduktegesetzes dienen der Erhebung von klinischen Daten, die zur Durchführung einer klinischen Bewertung erforderlich sind.
Eine Definition der klinischen Prüfung ist weder in der RL 93/42/EWG, noch im Medizinproduktegesetz und der Verordnung über die klinische Prüfung mit Medizinprodukten (MPKPV) zu finden.
Gemäß der DIN EN ISO 14155 ist die klinische Prüfung definiert als eine „systematische Prüfung an einer oder mehreren Versuchsperson(en), die vorgenommen wird, um die Sicherheit oder Leistungsfähigkeit eines Medizinprodukts zu bewerten“1.
Die klinische Prüfung ist in der Regel immer dann erforderlich, wenn sich im Rahmen der klinischen Bewertung herausstellt, dass nicht ausreichend klinische Daten aus der Literatur zur Verfügung stehen, um die Konformität mit den Grundlegenden Anforderungen (Anhang I RL 93/42/EWG) für das betreffende Medizinprodukt nachzuweisen.
Über die klinische Prüfung werden die noch fehlenden klinischen Daten für das betreffende Medizinprodukt erhoben, die zum Nachweis der Erfüllung der Grundlegenden Anforderungen notwendig sind.
Die klinische Prüfung verfolgt somit die gleichen Ziele wie die klinische Bewertung, nämlich
Im Medizinproduktegesetz ist die Erforderlichkeit der klinischen Prüfung allerdings nicht explizit geregelt.
Der Gesetzgeber verweist uns hier auf den Anhang X, Punkt 1.1.a der RL 93/42/EWG, welcher ausweist, dass klinische Prüfungen bei Produkten der Klasse III und implantierbaren Produkten durchzuführen sind.
Durch den Zusatz „es sei denn, die Verwendung bereits bestehender klinischer Daten ist ausreichend gerechtfertigt“2, besteht auch für diese Produkte nicht zwingend eine Verpflichtung eine klinische Prüfung durchzuführen.
Es gibt zahlreiche Regularien/Vorschriften, die sehr konkrete Vorgaben machen, wie eine klinische Prüfung mit einem Medizinprodukt durchzuführen ist. Dazu zählen:
In der EU-Richtlinie 93/42/EWG (vom 14.06.1993, zuletzt geändert über die Änderungs-Richtlinie 2007/47/EG, welche am 21.03.2010 in Kraft trat) wird der Rechtsrahmen über die allgemeinen Anforderungen an die Durchführung der klinischen Prüfung von Medizinprodukten im Artikel 15 mit den Anhängen VIII und X definiert.
In deutsches Recht umgesetzt sind diese Anforderungen aus der EU-Richtlinie 93/42/EWG im Medizinproduktegesetz (MPG) in den §§20 – 24. Durch die Novellierung des MPG über das Gesetz zur Änderung medizinprodukterechtlicher Vorschriften (4. MPG Novelle, in Kraft getreten am 21.03.2010), wurden die grundsätzlichen und formalen Anforderungen an die klinische Prüfung mit Medizinprodukten noch einmal verschärft und an das Arzneimittelgesetz (AMG) angeglichen. Die Paragrafen §§ 20 bis 22 entsprechen im Wesentlichen den §§ 40-42 AMG.
Über die Verordnung über die klinische Prüfung von Medizinprodukten (MPKPV) werden zusätzlich noch die relevanten Gesichtspunkte für das Genehmigungsverfahren bei der Behörde, das Verfahren bei der Ethikkommission und die Durchführung der klinischen Prüfung für alle Beteiligten konkretisiert.
Ergänzt wird dies durch die Medizinprodukte-Sicherheitsplanverordnung (MPSV), in die zum Schutz der Patienten der Begriff des schwerwiegenden unerwünschten Ereignisses aufgenommen wurde. Durch die Verordnung über das datenbankgeschützte Informationssystem über Medizinprodukte (DIMDIV) wird das zentrale elektronische Einreichungsverfahren für die erforderlichen Anträge bei den Behörden und Ethikkommissionen für klinische Prüfungen mit Medizinprodukten geregelt.
Die Norm über die Klinische Prüfung von Medizinprodukten an Menschen – Gute klinische Praxis (DIN EN ISO 14155, 2012) legt die formellen Anforderungen an die gute klinische Praxis für die Durchführung der klinischen Prüfung mit Medizinprodukten fest, um die Sicherheit und Leistungsfähigkeit zu gewährleisten. Sie gibt eine detaillierte Anleitung zu den wichtigsten Abläufen der klinischen Prüfung, beschreibt die Studiendokumente und die Aufgaben von Sponsor und Prüfer.
Des Weiteren muss jede klinische Prüfung mit einem Medizinprodukt, analog zur Arzneimittelprüfung, im Einklang mit der Deklaration von Helsinki (in der letzten vom Weltärztekongress geänderten Fassung) stehen, zum Schutz der an einer klinischen Prüfung teilnehmenden Personen.
Ob es sich um eine klinische Prüfung nach § 20-23a MPG handelt und die unter Punkt 2 genannten Regularien/Vorschriften für die klinische Prüfung bei Medizinprodukten gelten, hängt im Wesentlichen von der Art der klinischen Prüfung ab.
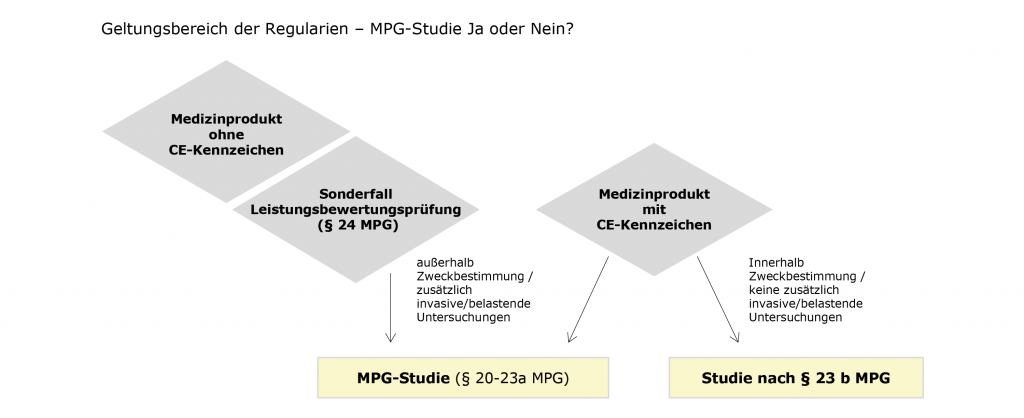
Sie gelten für:
Sie gelten nicht für:
Welche Art von Genehmigung für eine klinische Prüfung erforderlich ist, hängt in erster Linie von der Art der Studie ab. Für alle klinischen Prüfungen die unter das MPG (§20-23a) fallen, ist das Vorliegen einer Genehmigung durch die Bundesoberbehörde und die zustimmende Bewertung durch die Ethikkommission erforderlich, erst dann darf mit der klinischen Prüfung begonnen werden.
Das Antragsverfahren bei der Bundesoberbehörde und der Ethikkommission sind in den §§ 3-8 MPKPV geregelt. Bei MPG-Studien muss man hinsichtlich des Genehmigungsverfahrens bei der Bundesoberbehörde noch unterscheiden zwischen einem:
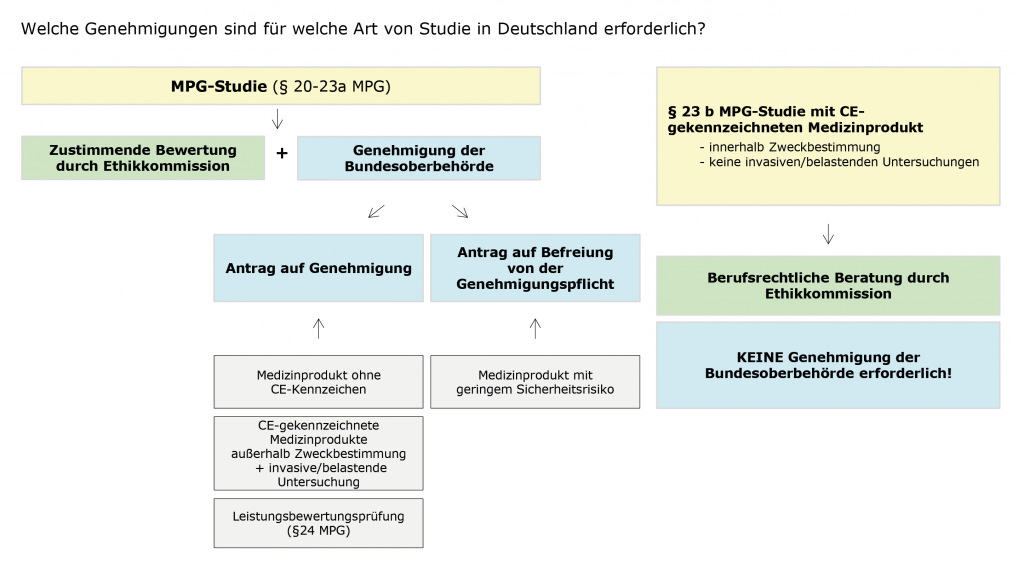
Antrag auf Genehmigung (gemäß § 3 MPKPV) ist bei der Bundesoberbehörde immer dann zu stellen, wenn es sich um eine MPG-Studie (§20-23a) handelt, die mit einem
Antrag auf Befreiung von der Genehmigungspflicht (gemäß § 7 MPKPV) ist bei der Bundesoberbehörde immer dann zu stellen, wenn es sich um eine MPG-Studie (§20-23a) mit einem Medizinprodukt mit geringem Sicherheitsrisiko handelt.
Antrag auf zustimmende Bewertung durch die Ethikkommission ist unabhängig davon, ob man einen Antrag auf Genehmigung oder einen Antrag auf Befreiung von der Genehmigungspflicht bei der Bundesoberbehörde stellen muss, immer erforderlich.
Für alle anderen Arten von Studien die sogenannten §23b –Studien bezeichnet werden, ist keine Genehmigung durch die Bundesoberbehörde und kein Ethikvotum erforderlich. Für die an solchen Studien teilnehmenden Ärzte gelten die berufsrechtlichen Vorgaben, wonach eine entsprechende Ethikkommission zur Beratung mit einzubeziehen ist (§ 15 BO Ärzte).
Bei einer klinischen Prüfung mit einem Medizinprodukt mit geringem Sicherheitsrisiko kann die Bundesoberbehörde von einer Genehmigung absehen. Dazu muss ein entsprechender Antrag bei der Bundesoberbehörde gestellt werden.
Der Antrag auf Befreiung von der Genehmigungspflicht ist immer dann möglich, wenn folgende Voraussetzungen für die klinische Prüfung gegeben sind: